миелодиспластический синдром что это такое прогноз
Миелодиспластический синдром у взрослых
Общая информация
Краткое описание
Приложение 5
к приказу
Министерства здравоохранения
Республики Беларусь
КЛИНИЧЕСКИМ ПРОТОКОЛ
диагностики и лечения пациентов в возрасте старше 18 лет
с миелодиспластическим синдромом
1. Клинический протокол диагностики и лечения пациентов с заболеванием «миелодиспластический синдром» (далее-МДС) предназначен для оказания медицинской помощи в амбулаторных и стационарных условиях районных, областных и республиканских организаций здравоохранения, имеющих в своем составе гематологические отделения.
2. Возрастная категория: взрослое население.

Автоматизация клиники: быстро и недорого!
— Подключено 300 клиник из 4 стран

Автоматизация клиники: быстро и недорого!
Мне интересно! Свяжитесь со мной
Классификация
КЛАССИФИКАЦИЯ ЗАБОЛЕВАНИЯ
5. Классификация МДС, принятая Всемирной организацией здравоохранения (далее-ВОЗ) в 2008 году базируется на цитоморфологических, кариотипических и клинических признаках заболевания.
Классификация миелодиспластических синдромов
сбалансированные аномалии: t(l 1; 16)(q23;p 13.3); t(3;21) (q26.2;q22.1); t(l;3) (рЗб.З; q21.1); t(2;l 1) (p21;q23); inv(3) (q21q26.2); t(6;9)(p23;q34);
сложный кариотип (3 или более хрмосомных аномалий) с вовлечением вышеупомянутых нарушений.
Диагностика
КРИТЕРИИ ДИАГНОЗА МДС
8. Вспомогательные критерии (С) (для пациентов, имеющих критерии А, но не имеющих критерии В).
8.1. Аномальный иммунный фенотип эритроидных или миелоидных клеток костного мозга, указывающий на их клональное происхождение (по результатам проточной цитометрии).
8.2. Молекулярно-генетические признаки наличия клональной клеточной популяции в костном мозге (по результатам HUMARA исследования или биологического микрочипирования).
8.3. Значительное и стабильное снижение колониеобразующей активности костного мозга и/или периферической крови.
Диагноз устанавливается при наличии 2 предварительных критериев (А) и не менее чем одного МДС-ассоциированных критериев (В). Вспомогательные
критерии (С) используются при отсутствии критериев В и наличии у пациента признаков клональной миелоидной пролиферации. Критерии группы С не входят в обязательный стандарт диагностики МДС.
Диагноз «идиопатическая цитопения неопределенного значения» применяется для обозначения случаев цитопении по одной и более клеточным линиям в течение > 6 месяцев при отсутствии критериев МДС и других причин цитопении. Такие пациенты должны наблюдаться и обследоваться гематологом с интервалом 1-6 месяцев.
Диагностические критерии разработаны ICWG (International Consensus Working Group), 2007 г.
Алгоритм диагностики МДС включает в себя клинические и лабораторные исследования, мультидисциплинарный подход с привлечением смежных специалистов и последовательно проводится на базе учреждений здравоохранения различного уровня с соблюдением преемственности на всех этапах. Это обусловлено полиэтиологичностью и гетерогенностью проявлений при данной патологии, стремлением к рационализации использования специального диагностического оборудования, минимизации диагностических ошибок.
Этапы диагностики МДС:
Лечение
КЛИНИЧЕСКИЕ ВАРИАНТЫ МДС
10. Определение клинического варианта МДС имеет значение для выбора тактики лечения.
10.1. 5ц-синдром: болеют преимущественно женщины, характерны вялотекущий характер заболевания, низкая вероятность трансформации в ОМЛ (10%), тяжелая макроцитарная анемия, нормальный или умерено сниженный уровень лейкоцитов и тромбоцитов, дисплазия мегакариоци- тарного ростка, отсутствие значительно повышения уровня бластных клеток в костном мозге; хороший ответ на леналидомид*.
10.2. Вторичный МДС: частота вторичного МДС нарастает в связи с успехами химиотерапии опухолей и воздействием загрязнения окружающей среды; для большинства пациентов характерны множественные хромосомные аберрации; прогноз хуже, чем при первичном МДС.
10.3. Гипопластический МДС:
до 15% случаев МДС характеризуются низкой клеточностью костного мозга при гистологическом исследовании (доля кроветворной ткани в препарате менее 30% у пациентов моложе 60 лет или менее 20% у пациентов 60 лет и старше);
дисплазия мегакариоцитов и клеток миелоидного ряда может отсутствовать;
возможны трудности в дифференциации от апластической анемии, для которой характерна более выраженная панцитопения, отсутствие типичных для МДС хромосомных аббераций и снижение содержания CD34+ клеток в костном мозге.
10.4. МДС с миелофиброзом: до 50% случаев всех вариантов МДС характеризуется фиброзом костного мозга (до 15% имеют выраженный фиброз); фиброз более характерен для вторичного МДС; характерны ги- перклеточность костного мозга, диффузный ретикулиновый фиброз его стромы и дисплазия не менее чем в 2 клеточных линиях; в периферической крови панцитопения, признаки клеточной дисплазии и лейкоэрит- робластоза; органомегалия нехарактерна; заболевание быстро прогрессирует; необходимо дифференцировать от острого мегакариобластного лейкоза, острого миелофиброза (острого панмиелоза с фиброзом), хронических миелопролиферативных заболеваний, метастатического рака, лимфом и волосатоклеточного лейкоза.
ЛЕЧЕНИЕ
11. Выбор терапии основан на диагнозе и группе риска по международной прогностической бальной системе (IPSS). В соответствии с международными рекомендациями для выбора терапевтической тактики пациентов с МДС подразделяют на 2 большие группы риска:
группу относительно низкого риска, включая в нее пациентов с низким и промежуточным 1 риском по системе IPSS;
группу высокого риска, включая в нее пациентов с промежуточным 2 и высоким риском по системе IPSS.
У пациентов из группы относительно низкого риска возможно применение только поддерживающей терапии либо терапии малой интенсивности. Интенсивная терапия показана пациентам группы высокого риска с учетом возраста, анамнеза заболевания, клинических проявлений, общего состояния и наличия признаков прогрессирования заболевания.
11.1. Поддерживающее лечение.
Поддерживающее лечение назначают с целью уменьшения проявлений заболевания и поддержания качества жизни. У пациентов из группы относительно низкого риска это может быть основным видом терапии.
11.1.1. Трансфузии донорских эритроцитов. Основным клиническим показанием для трансфузии донорских эритроцитов является не столько уровень гемоглобина, сколько степень адаптированности пациента к анемии.
11.1.2. Применение хелаторов железа.
Показаниями к применению хелаторов железа является переливание более 20-25 доз эритроцитной массы, уровень сывороточного ферритина более 2500 мкг/л, наличие дисфункции сердца (аритмия, сердечная недостаточность) и поражения печени.
Дефероксамин применяют в дозе 30-40 мг/кг в виде 12 часовых подкожных инфузий 5-7 раз в неделю (ночью). Дозу лекарственного средства снижают до 25 мг/кг при уровне ферритина ПО г/л, тромбоциты > 100 • 10 9 /л, нейтрофилы >1,0-10 9 /л, нет бластных клеток
время ремиссии, уменьшение абсолютного числа гранулоцитов, тромбоцитов, снижение концентрации НЬ на > 15 г/л или появление трансфузионной зависимости
Частичный: сокращение количества клеток, имеющих хромосомные аномалии на > 50 %
Гранулоциты (начальное количество 9 /л): прирост на 100 % и абсолютное количество > 0,5 • 10 9 /л
Прогноз
Для оценки прогноза и определения тактики лечения пациентов с МДС используют международную прогностическую бальную систему (далее IPSS).
Миелодиспластические синдромы
Миелодиспластические синдромы (МДС) объединяют группу злокачественных опухолевых заболеваний системы кроветворения.
При этих заболеваниях происходит нарушение созревания клеток костного мозга с изменением их строения и функциональных свойств.
Характерной, но не обязательной чертой заболевания является исход в острый лейкоз.
Эпидемиология
Заболеваемость МДС в среднем составляет 3-4 случая на 100 000 населения в год и увеличивается с возрастом.
Заболеваемость мужчин незначительно преобладает над заболеваемостью женщин. МДС в детском возрасте встречается крайне редко.
В 10-15% случаев МДС является осложнением проведенной химиотерапии и облучения по поводу другого онкологического заболевания.
Истинная заболеваемость МДС остается неизвестной. Связано это с бессимптомным течением заболевания у многих больных, трудностями диагностики и др. причинами.
Факторы риска
Известно, что факторы риска развития МДС и острых миелоидных лейкозов схожи.
В детском возрасте основными предрасполагающими факторами развития МДС являются генетические заболевания (синдромы Дауна, Фанкони, нейрофиброматоз, врожденный дискератоз и др.).
Генетическая предрасположенность отмечена и у лиц зрелого возраста, родители которых страдали МДС.
К химическим факторам риска относятся органические соединения (бензин и его производные, пестициды, растворители), а также неорганические (асбест, кварц, мышьяк) вещества.
МДС развивается чаще у курящих по сравнению с некурящими.
Более высокий риск возникновения МДС отмечен у работников сельского хозяйства, текстильной промышленности, лечебных учреждений, операторов машин, у лиц, проживающих вблизи заводов.
Наиболее изученным фактором риска является влияние химиопрепаратов и лучевого лечения. Чаще всего МДС диагностируется у больных, лечившихся по поводу рака молочной железы, множественной миеломы, лимфомы Ходжкина (лимфогранулематоза), неходжкинской лимфомы (лимфосаркомы), мелкоклеточного рака легкого.
Лучше всего МДС изучены у больных лимфомой Ходжкина. Частота развития МДС у этой категории больных колеблется от 1,5 до 10% и зависит от возраста, предшествующего лечения и длительности наблюдения.
Средний период от начала лечения болезни Ходжкина до установления диагноза МДС составляет около 4-6 лет. Убедительных данных об увеличении риска МДС при комбинации химиотерапии и облучения по сравнению с использованием только химиотерапии нет.
Клинические проявления
Признаки и симптомы у больных с МДС неспецифичны. У многих больных на протяжении ряда лет МДС протекает бессимптомно и выявляется лишь при обследовании по поводу других заболеваний.
Анемия (малокровие) является самым частым симптомом МДС и обнаруживается в 85-90% случаев. Большинство жалоб обусловлено выраженностью малокровия. Лейкопения (снижение числа лейкоцитов), отмечаемая у 50% больных, может быть причиной склонности к инфекционным осложнениям. В отдельных случаях повышение температуры может быть симптомом МДС. Иногда больные обращаются к врачу в связи со склонностью к кровотечениям из-за снижения числа тромбоцитов.
Несколько реже имеется увеличение лимфатических узлов. Увеличение, печени и лимфатических узлов при других вариантах МДС встречается редко.
Диагностика
Выявление МДС основывается на обнаружении признаков дисплазии (нарушений формирования ткани) в одном или нескольких ростках кроветворения.
При этом выявленные изменения могут быть чрезвычайно разнообразными. Соотношение нормальных и измененных клеток у разных больных существенно варьируют.
Принято считать клеточные линии (ростки кроветворения) измененными, если число клеток с признаками дисплазии составляет более 10%.
В том случае, если число измененных клеток в костном мозге невелико (менее 10%), для достоверной диагностики МДС необходимо проводить цитогенетическое исследование.
Прогностические факторы
Прогноз (исход) МДС зависит от многих факторов.
Продолжительность жизни и вероятность трансформации (перехода) в острый лейкоз при МДС существенно варьируются.
Самая короткая продолжительность жизни (5-7 месяцев) отмечается у больных МДС, вызванным химиотерапией и облучением. Кроме того, прогноз менее благоприятный у пациентов старше 60 лет, мужского пола и при наличии симптомов интоксикации.
Среди показателей крови к неблагоприятным прогностическим признакам относятся:
Наибольшее прогностическое значение имеет степень опухолевой инфильтрации (поражения) костного мозга. Прогноз менее благоприятный, если число опухолевых клеток в костном мозге превышает 10%. Некоторые выявленные хромосомные изменения также отрицательно сказываются на исходе заболевания.
Лечение
Существует два подхода к лечению МДС:
— радикальный, направленный на достижение и сохранение полной ремиссии,
— нерадикальный, направленный на улучшение качества жизни больных.
При нерадикальном подходе допускается существование (персистирирование) болезни, что принципиально отличает лечение МДС от терапии острых лейкозов.
Методы лечения МДС:
Выбор различных направлений терапии определяется: возрастом больного, наличием донора стволовых клеток, возможностью проведения адекватной сопроводительной терапии, а также эффективностью отдельных препаратов, подобранных с учетом исходных клинико-лабораторных показателей.
Практически с помощью каждого из перечисленных методов терапии возможно достижение полной ремиссии. Препараты, улучшающие качество жизни, успешно используются у больных с благоприятным прогнозом (с длительным сроком жизни и низкой вероятностью развития острого лейкоза).
При благоприятном прогнозе с минимальными проявлениями заболевания можно ограничиться наблюдением за больным, пока показатели крови и костного мозга остаются стабильными.
При выраженном малокровии показано переливание эритроцитарной массы. В ряде случаев необходимо применение ростовых факторов, антитимоцитарного глобулина, индукторов дифференцировки клеток и пр.
При прогрессировании МДС проводится химиотерапия.
У больных с неблагоприятным прогнозом проводится химиотерапия по программам лечения острого миелоидного лейкоза. При невозможности химиотерапии или высоком риске осложнений допустимо использование противоопухолевых препаратов в малых дозах.
Такой подход может позволить улучшить качество жизни больных и провести адекватное лечение в более поздние сроки.
После достижения полной ремиссии и при невозможности проведения аллогенной трансплантации костного мозга может осуществляться аутологичная трансплантация костного мозга или периферических стволовых клеток.
Необходимо подчеркнуть, что единственным методом лечения, позволяющим существенно увеличить продолжительность жизни больных МДС, является аллогенная трансплантация костного мозга или периферических стволовых клеток.
Однако применение аллогенной трансплантации не всегда возможно в связи с пожилым возрастом большинства больных и отсутствием идентичного родственного донора.
До настоящего времени результаты лечения больных МДС остаются неудовлетворительными.
В связи с этим практически не существует общепринятых стандартов лечения, а определены лишь общие подходы к терапии для разных групп больных при разных вариантах заболевания.
Мероприятия 2022 года
Архив мероприятий
Противораковое общество РОССИИ создано по инициативе ученых-онкологов и главных врачей онкологических диспансеров, представляющих более 50 регионов России, с целью претворения в жизнь программы профилактики рака в России
Миелодиспластический синдром
Миелодиспластический синдром относится к группе гематологических заболеваний, при которых нарушается нормальная работа органов кроветворения и изменяются основные показатели крови. Выявляется чаще у лиц трудоспособного возраста, у детей встречается редко. Особую актуальность для современной медицины миелодиспластический синдром приобрел, потому что он повышает риск развития лейкоза.
Виды миелодиспластического синдрома
В зависимости от факторов, которые послужили причиной развития заболевания, выделяют два основных вида миелодиспластического синдрома.
Существует более подробная классификация миелодиспластического синдрома, которая разработана экспертами ВОЗ. Она учитывает количество бластов в периферической крови, степень дисплазии костного мозга и общие показатели крови. В этой классификации выделяется форма миелодиспластического синдрома, которая ассоциирована с делецией длинного плеча 5-й хромосомы. Медико-генетический центр «Геномед» имеет необходимое оборудование для того, чтобы выявить данную мутацию.
Заболевание характеризуется нарушением кроветворения, поэтому клиническая картина будет зависеть от степени выраженности этих нарушений. Обычно для миелодиспластического синдрома характерны следующие проявления:
Если миелодиспластический синдром протекает в мягкой форме, то первые признаки могут появиться нескоро, а степень их выраженности обычно слабая. Длительное скрытое течение и стертая симптоматика не вызывает у пациентов настороженности и не заставляет обращаться за медицинской помощью, поэтому заболевание часто выявляется случайно.
Для того чтобы достоверно подтвердить миелодиспластический синдром, необходимо пройти комплексное гематологическое обследование, в которое включаются:
Хромосомные нарушения выявляются у 75 % больных, поэтому дополнительно назначается генетическое исследование.
Лечение миелодиспластического синдрома
План лечения составляется согласно тяжести заболевания и степени выраженности изменений в показателях крови. Если у пациента не выявляется анемия, геморрагические проявления и сопутствующие осложнения, то лечение в таких случаях не проводится, но необходимость в регулярном контроле и наблюдении сохраняется. Если же нарушения выражены существенно, выявлен высокий риск развития лейкоза, то назначается терапия, направленная на подавление иммунных реакций, возможна химиотерапия, регулярные переливания крови и ее компонентов, в тяжелых случаях пересадка костного мозга.
Продолжительность жизни пациентов с миелодиспластическим синдромом зависит от клинических проявлений и успешности лечения. В лучшем случае она составляет 10 лет и более, а в самых худших – меньше года.
Миелодиспластический синдром что это такое прогноз
Миелодиспластические синдромы гетерогенны по продолжительности жизни и вероятности трансформации в острый лейкоз. Прогноз течения миелодиспластического синдрома (МДС) определяется как особенностями биологии заболевания, возрастом больного, его общим состоянием, наличием сопутствующих заболеваний, так и выбором терапии.
Вполне понятно, что при использовании разных препаратов или схем лечения миелодиспластического синдрома (МДС) выявлены различные прогностические факторы, влияющие на успех терапии. В связи с этим в работах, направленных на поиск прогностической роли клинико-лабораторных симптомов миелодиспластического синдрома (МДС), используются выборки больных, получавших либо только симптоматическую терапию, либо лечение, которое не влияет на продолжительность жизни и вероятность трансформации заболевания в ОМЛ.
Среди клинических признаков прогностическое значение имеют пол и возраст. Продолжительность жизни больше у больных моложе 60 лет и у женщин. На вероятность трансформации в острый лейкоз пол и возраст влияния не оказывают. Отрицательную прогностическую роль играет наличие симптомов интоксикации. Особенно короткая выживаемость отмечена при вторичных миелодиспластических синдромов (МДС) (5— 7 мес).
Прогностическое значение ФАБ-вариантов миелодиспластических синдромов (МДС)

Среди показателей крови к неблагоприятным прогностическим факторам относятся: анемия, нейтропения и тромбоцитопения. Негативное влияние на течение миелодиспластического синдрома (МДС) оказывает не только наличие цитопении разной степени выраженности, но и ее характер (одно-, двух- или трехростковый). Трехростковая цитопения рассматривается как несомненно неблагоприятный признак. К неблагоприятным прогностическим признакам относится наличие в крови 5 % и более бластных клеток, нормоцитов и молодых форм нейтрофилов, а также повышенный уровень ЛДГ.
Среди показателей костного мозга наиболее прогностически значимым считается число бластных клеток. Показано, что при числе бластных клеток более 10 % прогноз хуже, чем при их количестве 5— 10 %. Негативное прогностическое значение имеет большое число пельгероидных форм гранулоцитов и микроформ мегакариоцитов, наличие в трепанобиоптате скоплений бластных клеток, фиброза костного мозга, АЛМП.
Продолжительность жизни больных миелодиспластическим синдромом с различными изменениями кариотипа
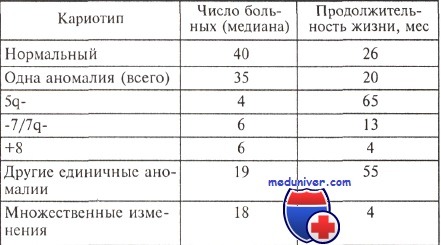
По данным G. Tricot и соавт., в группе больных с числом бластных клеток в миелограмме менее 5 % с выявленной АЛМП медиана выживаемости составила 14 мес, в то время как при отсутствии АЛМП — 49 мес. Некоторые авторы считают, что при низкой клеточности трансформация в острый лейкоз наступает реже и выживаемость больных выше. В исследовании A. Georgii и соавт. при гипопластическом варианте миелодиспластического синдрома (МДС) медиана выживаемости составила 18,6 мес., при других вариантах — 15,4 мес.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Миелодиспластический синдром

Миелодиспластический синдром – группа гематологических заболеваний, при которых наблюдаются цитопения, диспластические изменения костного мозга и высокий риск возникновения острого лейкоза. Характерные симптомы отсутствуют, выявляются признаки анемии, нейтропении и тромбоцитопении. Диагноз устанавливается с учетом данных лабораторных анализов: полного анализа периферической крови, гистологического и цитологического исследования биоптата и аспирата костного мозга и т. д. Дифференциальный диагноз может представлять значительные затруднения. Лечение – переливание компонентов крови, химиотерапия, иммуносупрессивная терапия, пересадка костного мозга.

Общие сведения
Миелодиспластический синдром – группа заболеваний и состояний с нарушениями миелоидного кроветворения и высоким риском развития острого лейкоза. Вероятность развития увеличивается с возрастом, в 80% случаев данный синдром диагностируется у людей старше 60 лет. Мужчины страдают несколько чаще женщин. У детей миелодиспластический синдром практически не встречается. В последние десятилетия гематологи отмечают увеличение заболеваемости среди лиц трудоспособного возраста. Предполагается, что причиной «омоложения» болезни могло стать существенное ухудшение экологической обстановки.
До недавнего времени лечение миелодиспластического синдрома было только симптоматическим. Сегодня специалисты разрабатывают новые методы терапии, однако эффективное лечение этой группы болезней все еще остается одной из самых сложных проблем современной гематологии. Пока прогноз при миелодиспластическом синдроме, в основном, зависит от особенностей течения болезни, наличия или отсутствия осложнений. Лечение осуществляют специалисты в сфере онкологии и гематологии.
Причины и классификация миелодиспластического синдрома
С учетом причин развития различают два типа миелодиспластического синдрома: первичный (идиопатический) и вторичный. Идиопатический вариант выявляется в 80-90% случаев, диагностируется преимущественно у пациентов старше 60 лет. Причины возникновения установить не удается. В числе факторов риска первичного миелодиспластического синдрома – курение, повышенный уровень радиации при выполнении профессиональных обязанностей или проживании в неблагоприятной экологической зоне, частый контакт с бензином, пестицидами и органическими растворителям, некоторые наследственные и врожденные заболевания (нейрофиброматоз, анемия Фанкони, синдром Дауна).
Вторичный вариант миелодиспластического синдрома наблюдается в 10-20% случаев, может возникать в любом возрасте. Причиной развития становится химиотерапия или радиотерапия по поводу какого-то онкологического заболевания. В число лекарственных средств с доказанной способностью вызывать миелодиспластический синдром включают циклофосфан, подофиллотоксины, антрациклины (доксорубицин) и ингибиторы топоизомеразы (иринотекан, топотекан). Вторичный вариант отличается более высокой резистентностью к лечению, более высоким риском развития острого лейкоза и более неблагоприятным прогнозом.
В современной редакции классификации ВОЗ различают следующие типы миелодиспластического синдрома:
Симптомы миелодиспластического синдрома
Клиническая симптоматика определяется степенью нарушений миелопоэза. При мягко протекающих расстройствах возможно длительное бессимптомное или стертое течение. Из-за слабой выраженности клинических проявлений некоторые больные не обращаются к врачам, и миелодиспластический синдром обнаруживается во время проведения очередного медицинского осмотра. При преобладании анемии наблюдаются слабость, одышка, плохая переносимость физических нагрузок, бледность кожных покровов, головокружения и обморочные состояния.
При миелодиспластическом синдроме с тромбоцитопенией возникает повышенная кровоточивость, отмечаются десневые и носовые кровотечения, на коже появляются петехии. Возможны подкожные кровоизлияния и меноррагии. Миелодиспластический синдром с выраженными нейтропенией и агранулоцитозом проявляется частыми простудами, стоматитом, синуситом или стрептодермией. В тяжелых случаях возможно развитие пневмонии или сепсиса. Инфекционные заболевания нередко вызываются грибками, вирусами или условно-патогенными микробами. У каждого пятого пациента с миелодиспластическим синдромом выявляется увеличение лимфоузлов, селезенки и печени.
Диагностика миелодиспластического синдрома
При исследовании костного мозга количество клеток обычно нормальное либо повышенное. Уже на ранних стадиях обнаруживаются признаки дизэритропоэза. Количество бластов зависит от формы миелодиспластического синдрома, может быть нормальным либо увеличенным. В последующем наблюдаются дисгранулоцитопоэз и дисмегакариоцитопоэз. У некоторых больных признаки дисплазии костного мозга выражены очень слабо. В процессе цитогенетического исследования у ¾ больных выявляются хромосомные нарушения. Дифференциальный диагноз миелодиспластического синдрома проводят с В12-дефицитной анемией, фолиево-дефицитной анемией, апластической анемией, острым миелолейкозом и другими острыми лейкозами.
Лечение и прогноз при миелодиспластическом синдроме
Тактика лечения определяется выраженностью клинической симптоматики и лабораторных изменений. При отсутствии явных признаков анемии, геморрагического синдрома и инфекционных осложнений осуществляется наблюдение. При миелодиспластическом синдроме с выраженной анемией, тромбоцитопенией и нейтропенией, а также при высоком риске возникновения острого лейкоза назначают сопроводительную терапию, химиотерапию и иммуносупрессивную терапию. При необходимости осуществляют пересадку костного мозга.
Сопроводительная терапия является самым распространенным методом лечения миелодиспластического синдрома. Предусматривает внутривенные инфузии компонентов крови. При длительном применении может провоцировать повышение уровня железа, влекущее за собой нарушения деятельности жизненно важных органов, поэтому переливания гемокомпонентов производят при одновременном приеме хелаторов (лекарственных средств, связывающих железо и способствующих его выведению).
Иммуносупрессоры эффективны при лечении миелодиспластического синдрома с отсутствием хромосомных аномалий, наличием гена HLA-DR15 и гипоклеточном костном мозге. Химиотерапию применяют при невозможности трансплантации костного мозга. Высокие дозы препаратов используют при трансформации миелодиспластического синдрома в острый лейкоз, а также при рефрактерных анемиях с избытком бластов при нормоклеточном и гиперклеточном костном мозге, низкие – при невозможности пересадки костного мозга. Наряду с перечисленными средствами пациентам назначают гипометилирующие средства (азацитидин). Наиболее надежным способом достижения полноценной длительной ремиссии является трансплантация костного мозга.
Прогноз зависит от типа миелодиспластического синдрома, количества хромосомных аномалий, необходимости в регулярных переливаниях компонентов крови, выраженности клинических проявлений и наличия осложнений. Различают 5 групп риска. Средняя выживаемость больных миелодиспластическим синдромом, входящих в группу с самым низким уровнем риска, составляет более 11 лет; с самым высоким – около 8 месяцев. Вероятность отторжения костного мозга после трансплантации – около 10%.